

 Print
Print facebook
facebook twitter
twitter Linkedin
Linkedin google+
google+

Introduction
Prenatally detected abdominal and pelvic masses are usually cystic in nature, and are typically seen on second-trimester sonography [1]. These lesions commonly arise from various organs and structures in the abdomen and pelvis [2]. Prenatal sonography is highly sensitive in the detection of abdominopelvic cystic masses, but it lacks specificity and a definitive diagnosis is often challenging [1-3]. Postnatal sonography is often successful in determining the etiology of cystic lesions, occasionally supplemented by magnetic resonance imaging (MRI). Sonography is the preferred modality in this setting, given its portability, lack of ionizing radiation, low cost, and high resolution in small patients. The differential diagnosis of abdominopelvic cystic masses in newborns is broad and can be narrowed by approaching the lesions based on their anatomic location: intraperitoneal, porta hepatis/hepatic, renal/retroperitoneal, splenic, or pelvic. In addition, distinctive sonographic signs, when present, aid in establishing the correct diagnosis [1,3]. An accurate diagnosis is pivotal, as the management of these cystic lesions is highly variable [2,4].
Intraperitoneal Cystic Masses
Ovarian Cysts
Ovarian cysts are the most common abdominal masses found in female neonates [5]. Neonatal ovarian cysts are primarily of follicular origin, presumably arising from disordered folliculogenesis. Excessive stimulation of the fetal ovary by both placental and maternal hormones is thought be a key factor in cyst development [6]. The decrease in hormonal stimulation after birth is associated with spontaneous regression of most small cysts; however, 5-cm-diameter or larger cysts are associated with an increased risk of torsion. There is an increased incidence of cysts in infants of mothers with diabetes, toxemia, or rhesus immunization, presumably from hypersecretion of placental human chorionic gonadotropin (hCG) or increased placental permeability to hCG [7].
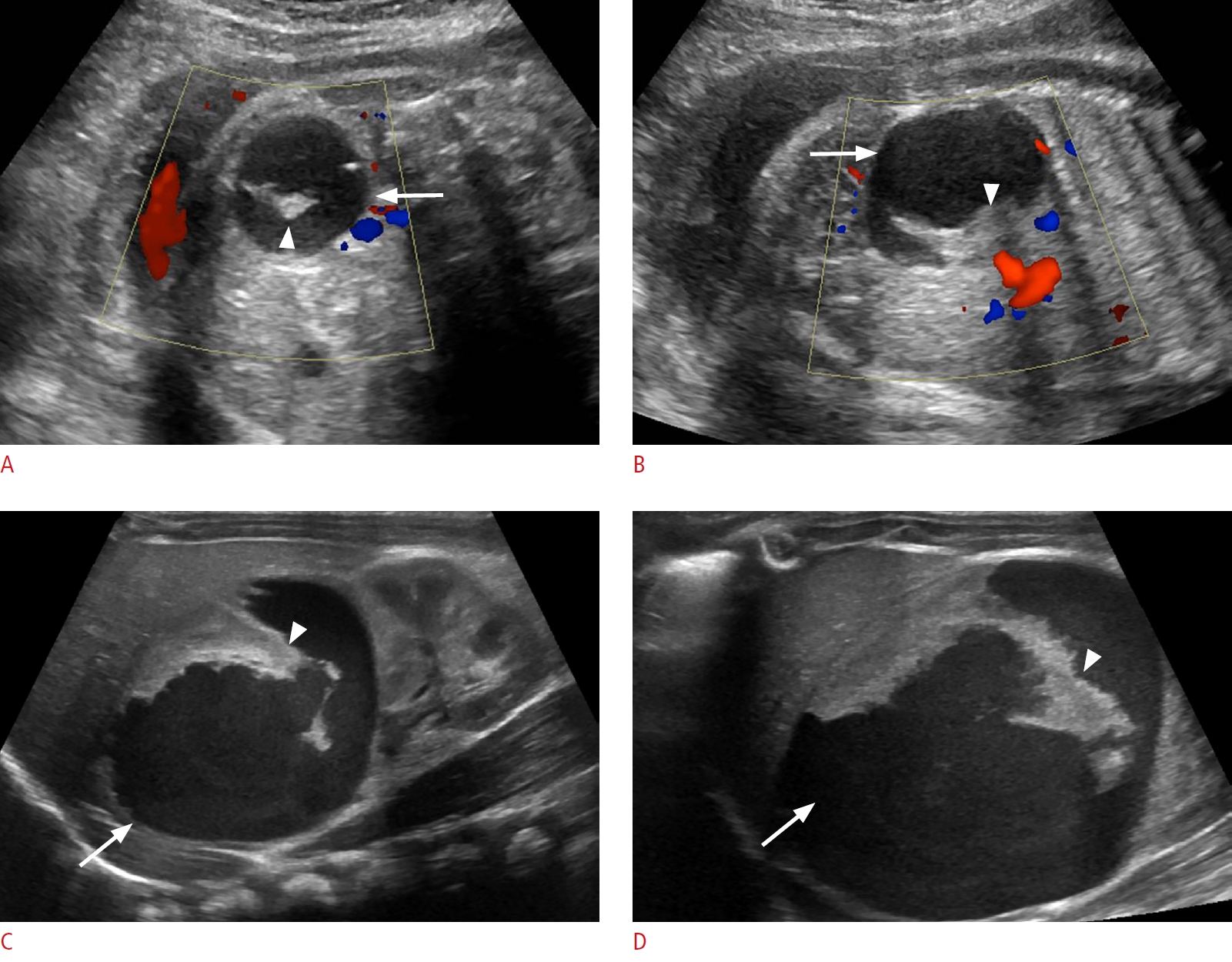
Neonatal ovarian cysts are categorized as simple or complicated; the latter type occurs in the setting of torsion, and mostly develops antenatally. They are most often unilateral, found anywhere within the peritoneal cavity in neonates, even contralaterally, due to the long neonatal ovarian pedicle. Larger cysts may occupy almost the entire abdomen [5,8-10].
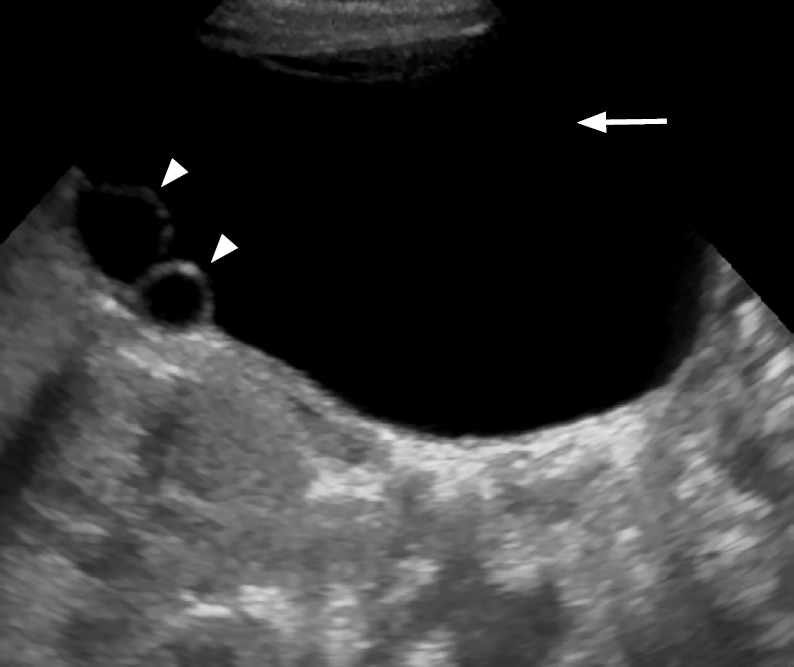
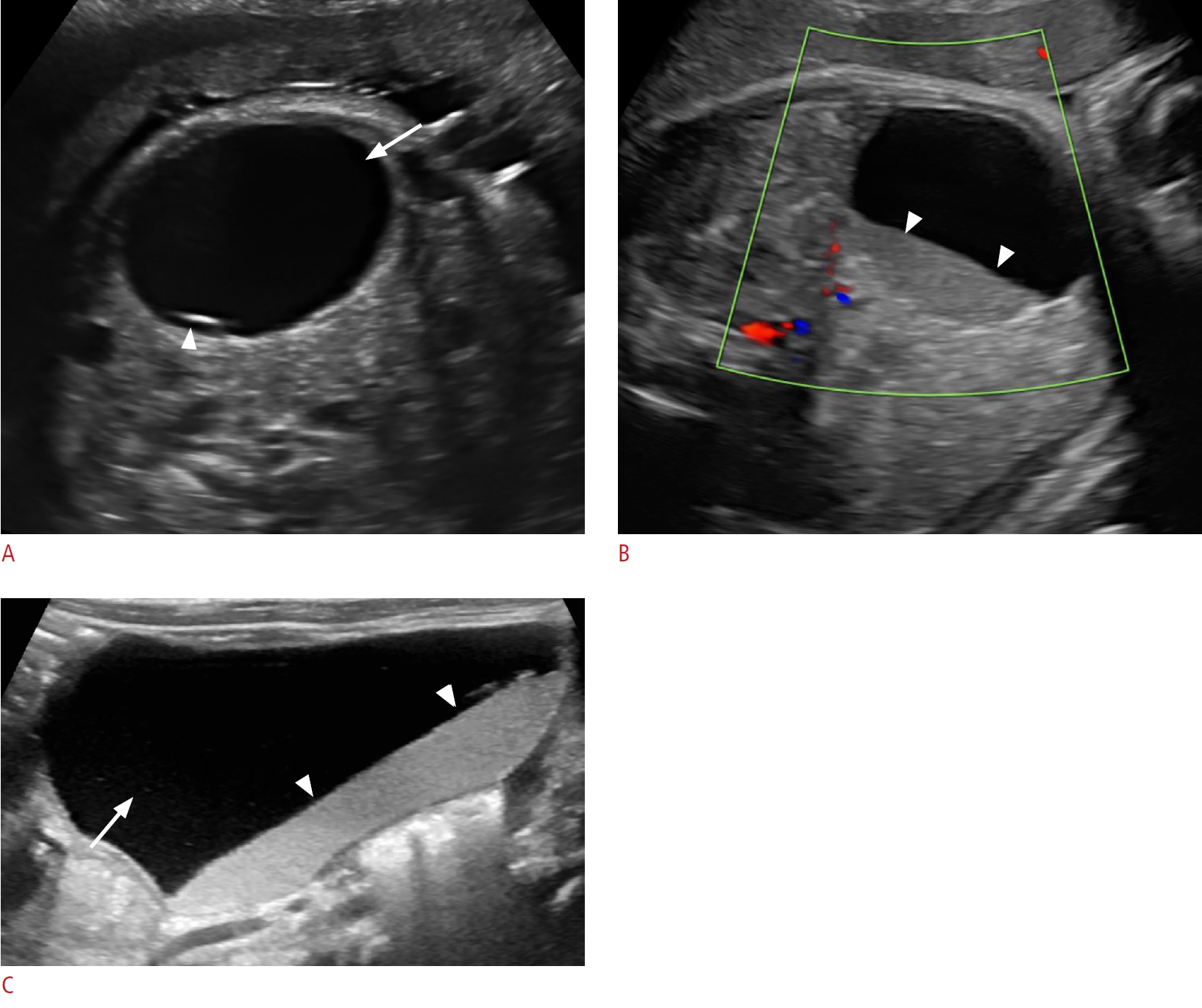
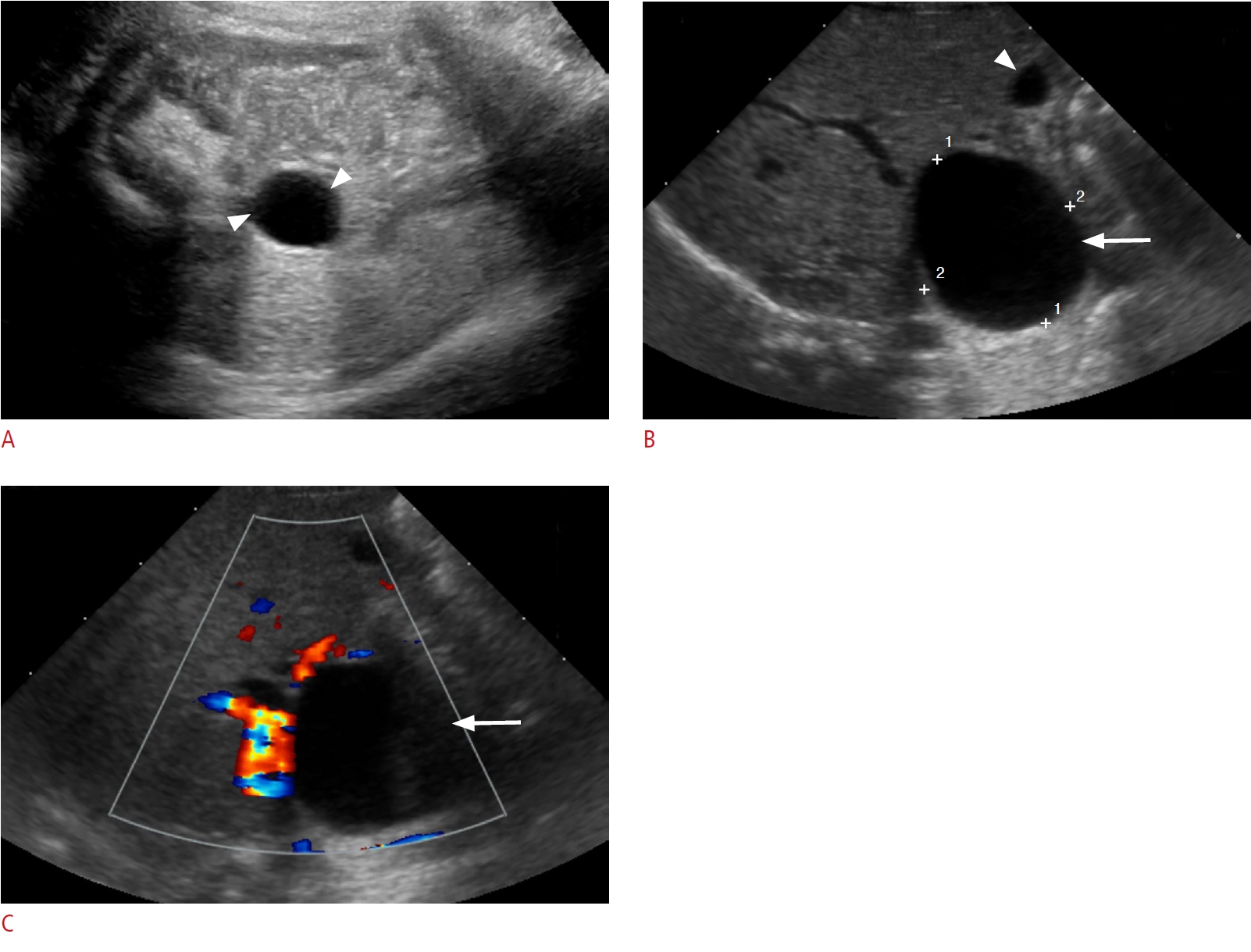
On sonography, simple ovarian cysts are unilocular, round, and anechoic with a thin wall, possibly with a single septum. The "daughter cyst" sign (Figs. 1, 2A), which is highly specific for ovarian cysts, denotes a small round thin-walled anechoic lesion abutting the wall of the ovarian cyst [5]. Ovarian torsion has a variable sonographic appearance related to degree of internal hemorrhage, stromal edema, and infarction. Ovarian torsion invariably contains a fluid-debris level, retracting clot, septations, or an area completely filled with echoes producing a solid mass-like appearance (Figs. 2B, C, 3). These complex cysts often have echogenic walls resulting from dystrophic calcification associated with infarction. The absence of internal blood flow on color Doppler evaluation distinguishes solid-appearing ovarian torsion from a rare solid intraperitoneal mass [5,8,11,12].
Complications of large ovarian cysts include rupture causing hemorrhagic ascites, peritonitis, or bowel obstruction; thoracic compression resulting in pulmonary hypoplasia; urinary tract obstruction; and incarceration of the ovarian mass within an inguinal hernia [7]. Most small simple ovarian cysts resolve spontaneously. Simple ovarian cysts more than 5 cm in diameter should be closely monitored with serial ultrasonography. Ovarian torsion merits careful assessment and surgical consultation; if surgical intervention is required, the goal should be to preserve ovarian tissue. Conservative management of complex ovarian cysts has been advocated if the neonate is stable [5,13,14].
Enteric Duplication Cysts
Enteric duplication cysts are congenital cysts of unknown etiology, arising anywhere along the gastrointestinal tract from the mouth to anus. These cysts are contiguous with a segment of bowel, sharing a muscular layer and arterial blood supply with the adjacent bowel. In the abdomen, they are most common in the ileocecal region, arising on the mesenteric border of the bowel. Most duplication cysts do not communicate with the intestinal lumen, except in the tubular type [5,15-17]. Ectopic gastric mucosa can be present in some cases which can present with intralesional hemorrhage [18].
On sonography, most enteric duplication cysts are round, and the tubular type accounts for 20%. They are anechoic with a thick well-defined wall. They have a distinctive "gut signature" or "double wall" sign, which represents the inner hyperechoic mucosal layer and the peripheral hypoechoic muscular wall (Fig. 4). At times, five layers of the gut signature (echogenic mucosa, hypoechoic muscularis mucosa, echogenic submucosa, hypoechoic muscularis propria and echogenic serosa) can be visualized with high-frequency transducers [17]. Rarely, the double wall sign can be seen in ovarian cysts, Meckel diverticula, lymphatic malformations (LMs), or cystic teratomas [18]. Lack of the double wall sign has been reported in duplication cysts, and thought to be related to cyst infection and erosion of the mucosa [17]. Identification of the "split wall" sign, which consists of hypoechoic muscularis propria divided at the point of attachment between the cyst and adjacent bowel, or peristalsis of the cyst wall increases specificity in establishing this diagnosis. Septations, internal echoes, or debris related to proteinaceous material, blood products, or infection can be present [5,17]. Duplication cysts can lead to intussusception and segmental volvulus; therefore, the treatment is surgical resection [5,16].
Meconium Pseudocysts
Meconium pseudocysts are rare in neonates, and often coexist with meconium peritonitis, which occurs in the setting of intrauterine bowel perforation. Prenatal bowel perforation can be caused by intestinal atresia, segmental bowel volvulus, meconium ileus, or spontaneous perforation. Leakage of bowel content into the peritoneum causes severe inflammation with adhesions and may result in the development of pseudocysts, reflecting the normal healing process to confine the intraabdominal perforation [5,10,19].
On sonography, meconium pseudocysts appear cystic, with a thick echogenic calcified wall, variable internal echogenicity, and absent vascularity (Fig. 5). Intraluminal gas can be seen if communication persists between the cyst cavity and the perforated bowel lumen after birth. Dilated bowel loops and ascites may be present [5,19-23]. Patients with complicated meconium pseudocysts are managed surgically [24]. In some patients, the perforated bowel spontaneously heals in utero, and the only clue of previous perforation is the finding of peritoneal calcifications [18].
Lymphatic Malformations
LMs consist of dilated lymphatic channels and spaces with walls lined by mature endothelium. The etiology is unknown; hypotheses include failure of embryonic lymphatic spaces to develop normal connections and drainage into the venous system, and benign proliferation of ectopic lymphatics sequestered from the venous system [25]. LMs have no sex predilection. The head and neck are the commonest sites of involvement, while the abdomen is less frequently involved, and these malformations can arise from small bowel mesentery, omentum, and retroperitoneum [26-28]. They can be asymptomatic or manifest with bowel obstruction, segmental volvulus, hemorrhage, rupture, infection, and torsion of the lesion [29].
LMs consist of cystic structures of variable size. The macrocystic type usually occurs in the abdomen. On sonography, macrocystic lesions appear as thin-walled, unilocular or multilocular anechoic lesions, often with fine septations. Internal echoes or fluid levels can be seen related to hemorrhage or infection. On color Doppler imaging, no flow is detected, although septal vascularity can be visualized because they often encase the neighboring mesenteric vessels. Ascites, often chylous, can be present [18,25,27,30]. Treatment may include percutaneous sclerotherapy, surgical resection, or a combination of both. The prognosis is generally good, with a low recurrence rate [26,28].
Urachal Cysts
Urachal cysts are part of a spectrum of congenital anomalies arising from incomplete obliteration of the urachus. The urachus connects the dome of the bladder to the umbilical cord during fetal life and is located between the parietal peritoneum and fascia transversalis (space of Retzius). Urachal cysts develop when both the umbilical and bladder end of the urachus are obliterated, but a focal segment remains patent along the course of the urachus. They are rare and more common in males than females, and they can be asymptomatic or manifest clinically with infection.
Uncomplicated urachal cysts appear as simple anechoic lesions on sonography located at the midline behind the lower anterior abdominal wall, in the space of Retzius, between the umbilicus and the bladder. Infected urachal cysts may have a complex sonographic appearance. Complete excision of the urachal remnant is the recommended treatment [31,32].
Porta Hepatis and Hepatic Cystic Masses
Choledochal Cysts
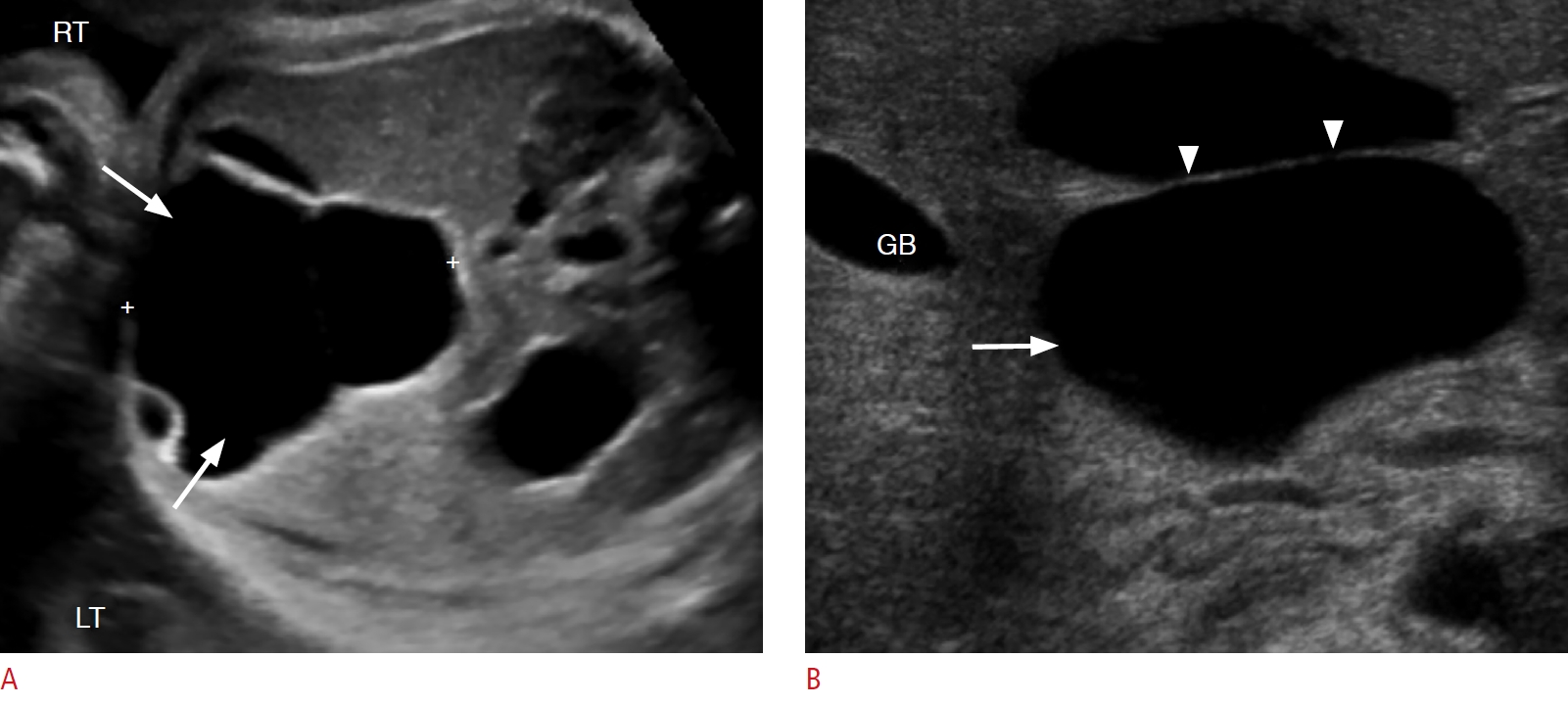
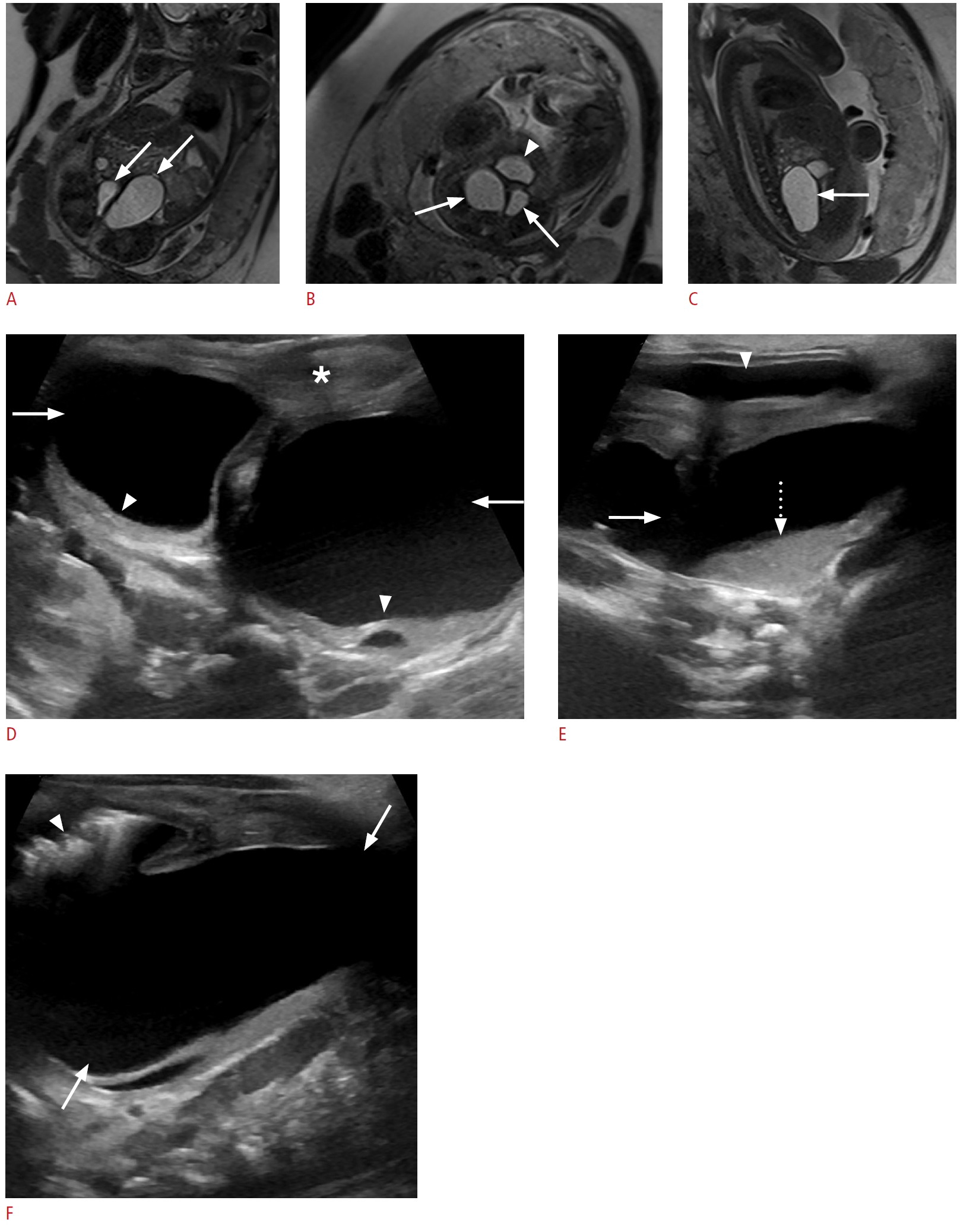
Choledochal cysts, which are fusiform or saccular dilatations of the bile ducts, are among the most frequent congenital hepatobiliary anomalies [33,34]. The Todani classification system is used to categorize these anomalies into five types based on their etiology, pathogenesis, appearance, and presentation [35]. Type I choledochal cysts are postulated to arise from ductal plate malformation and obstruction of the distal biliary duct and/or reflux of the pancreatic enzymes into the biliary tree, due to an anomalous pancreatobiliary junction [18]. A round or tubular cystic right upper quadrant mass separate from the gallbladder is seen in type I, II, and IV choledochal cysts on sonography (Fig. 6). Type III cysts (choledochocoele) may cause a mass effect at the ampulla of Vater. Multiple intrahepatic cystic dilatations are seen in type IVA and type V cysts. A normal gallbladder is present, usually adjacent to the dilated common duct (Fig. 6B). Sludge or stones may be seen in the dilated ducts. Choledochal cysts can be differentiated from cystic biliary atresia on sonography on the basis of gallbladder abnormalities, non-dilated bile ducts, smaller cyst (20 mm or less), and lack of sludge or stones, which are characteristic features of biliary atresia [5,34,36-38].
MRI with cholangiopancreatography is the modality of choice to delineate the type of cyst, length of duct involved, presence and location of protein plugs or calculi, and the length of the common channel [34,36]. Complications may include calculi, cholangitis, pancreatitis, or cholangiocarcinoma. Surgical excision with hepaticojejunostomy on an elective basis is the definitive treatment [5,36].
Cystic Biliary Atresia
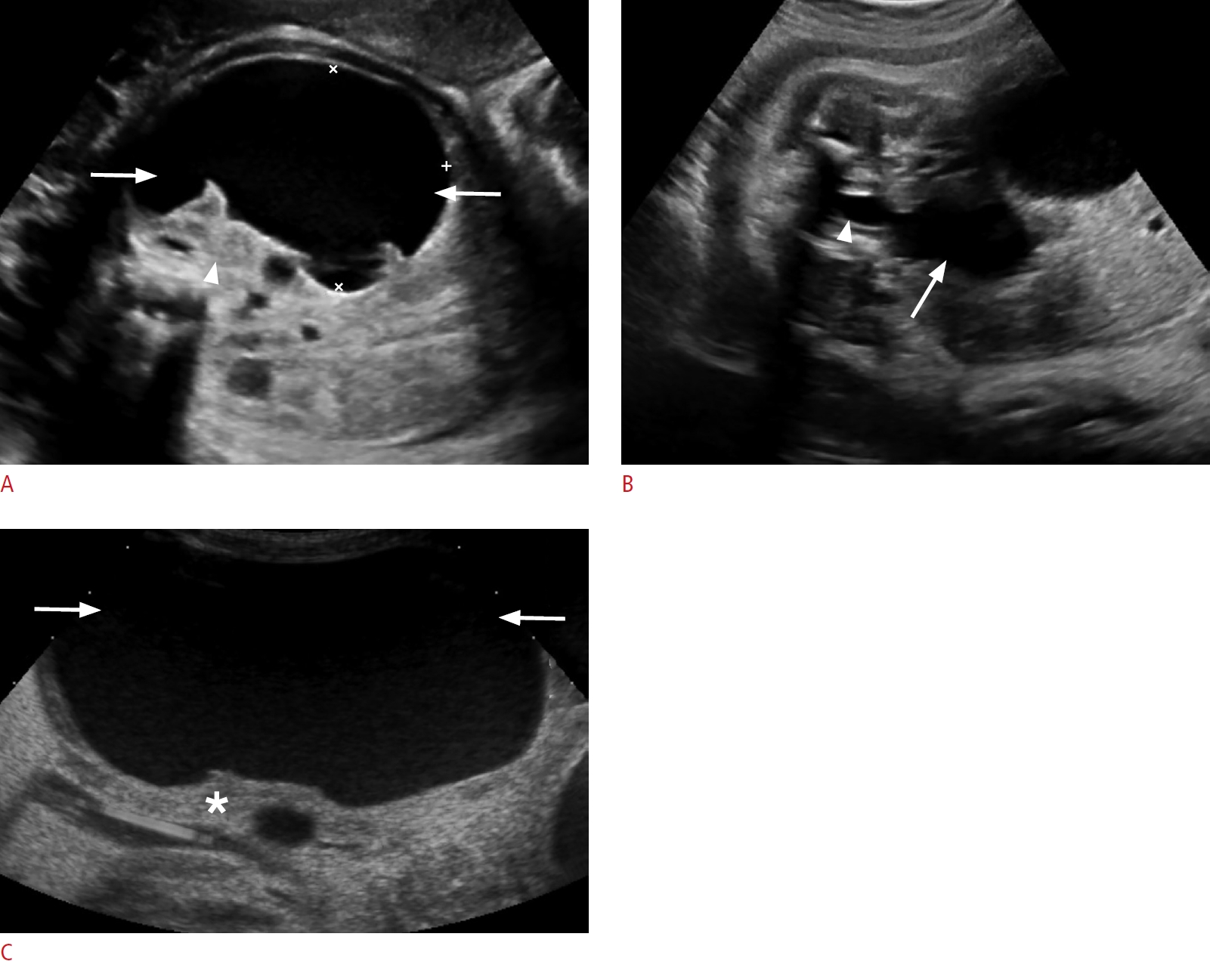
Cystic biliary atresia is a rare variant of biliary atresia presenting with a cystic structure within the otherwise obliterated extrahepatic bile duct. Biliary atresia is an inflammatory fibro-obliterative disease that affects varying lengths of both intrahepatic and extrahepatic bile ducts. It is a common cause of neonatal cholestasis and a common indication for pediatric liver transplantation. Biliary atresia is more prevalent in Asian populations [38,39]. The etiology and pathogenesis of biliary atresia remain unclear, although viral, genetic, toxic, and autoimmune origins have been proposed as possibilities [39]. Most cases of biliary atresia are isolated; however, about 10%-35% are associated with congenital anomalies such as situs inversus or polysplenia [38,40,41].
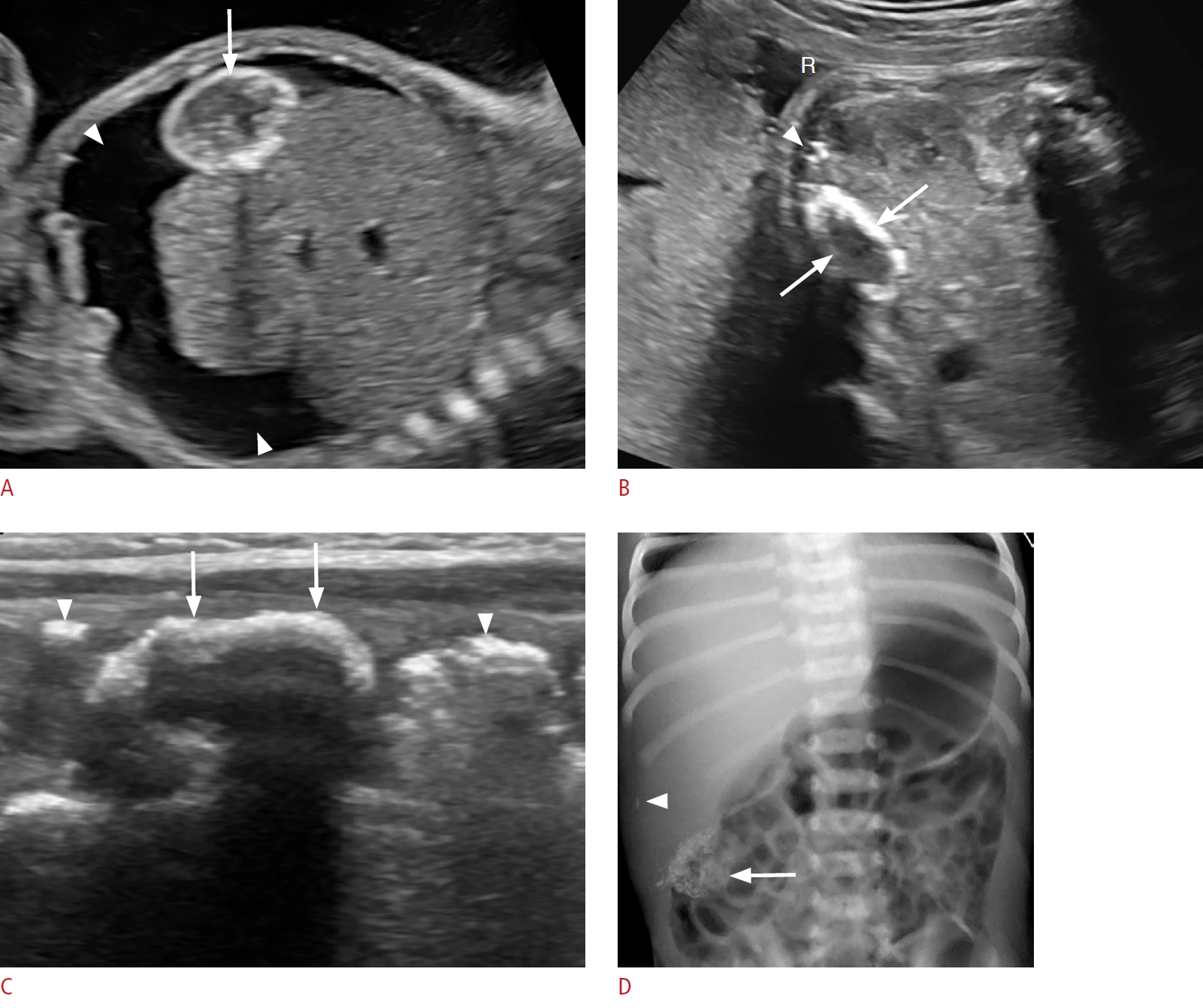
On sonography, cystic biliary atresia presents with a small cyst (20 mm or less) located at the porta hepatis (Fig. 7A-D). The cyst size tends to remain stable, whereas choledochal cysts may increase in size. Gallbladder abnormalities, such as absent gallbladder, small gallbladder (length <15 mm) or irregular gallbladder wall, and the triangular cord sign (>4 mm of echogenic tissue anterior to the portal vein) are each highly specific for the diagnosis of cystic biliary atresia on sonography (Fig. 7E, F). Absence of the common bile duct, non-dilated bile ducts, enlargement of the hepatic artery (>1.5 mm) and the presence of hepatic subcapsular flow are other important findings. Diagnostic accuracy for biliary atresia can be improved to 98% when the findings are combined [38,41-45]. The diagnosis of biliary atresia can be confirmed with an intraoperative cholangiogram and liver biopsy [40,41,46]. Cystic biliary atresia must be differentiated from choledochal cyst, as these two entities have dramatically different management approaches and prognoses. If left untreated, the former progresses to end-stage liver disease and death within the first 3 years of life [39]. The treatment of cystic biliary atresia is urgent surgery with the Kasai procedure [47].
Congenital Simple Hepatic Cysts
Congenital simple hepatic cysts are rare. These cysts are postulated to arise from in utero aberrant development of the biliary tree with obstruction resulting in fluid accumulation, ductule dilatation, and loss of communication with the normal biliary tree. They may also occur after obstruction of the peribiliary glands, either from a congenital malformation or another disease process [48,49]. There is no lobar propensity and only a slight female predominance [48-50].
On sonography, simple hepatic cysts typically present as solitary, simple, anechoic lesions of variable size within the hepatic parenchyma, usually with a lack of communication with the biliary tree (Fig. 8). Less commonly, they may be large and multilocular. Exophytic simple hepatic cysts may mimic choledochal cysts and cystic biliary atresia; however, hepatic cysts typically present with a normal gallbladder and normal size of the bile ducts and do not connect with the biliary tree. Hepatic cysts are usually not associated with other congenital anomalies [49-52]. They tend to remain stable in size or regress over time. Surgical treatment is reserved for symptomatic or enlarging lesions [48,51-54].
Mesenchymal Hamartoma
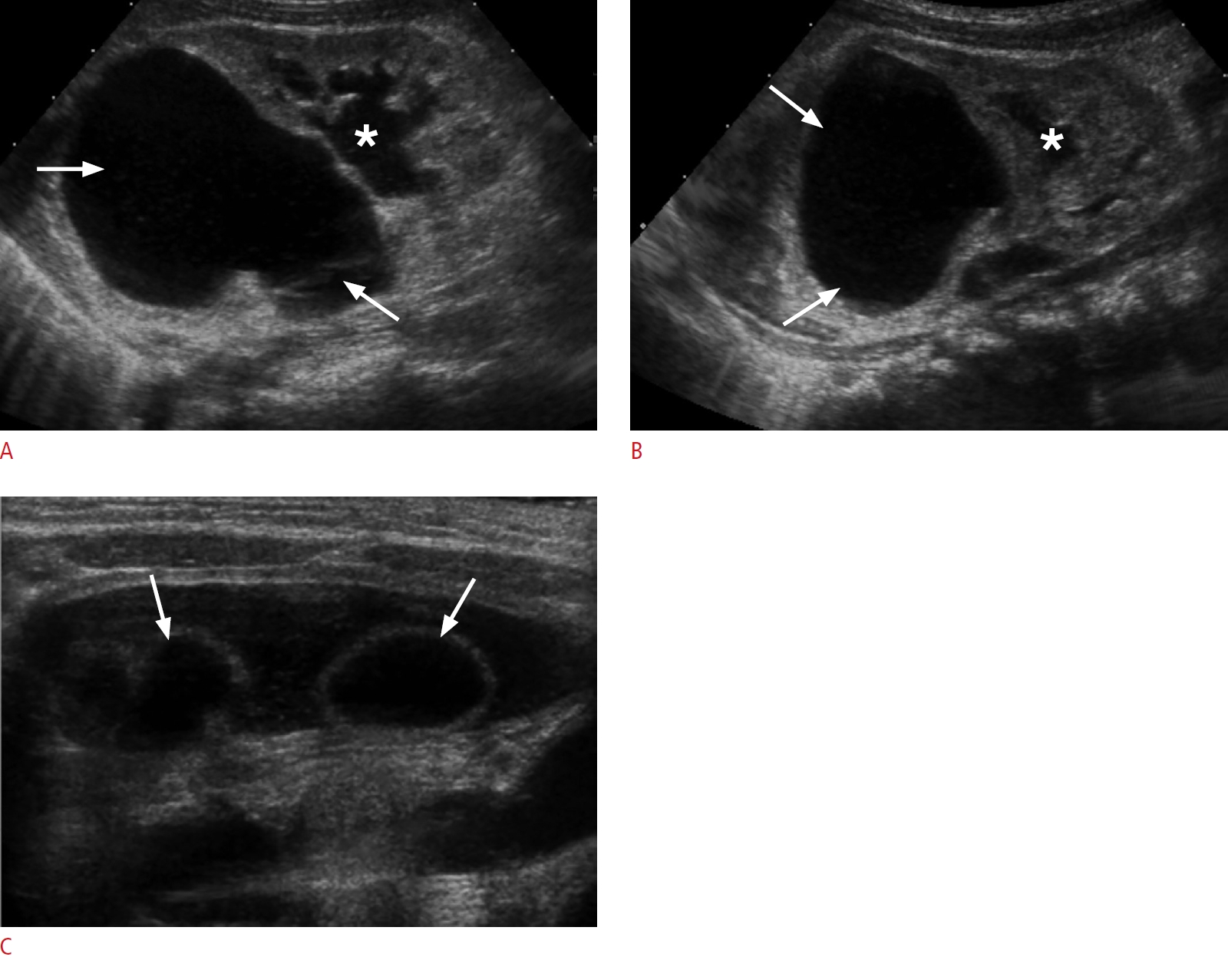
Mesenchymal hamartoma is one of the most common benign liver tumors in children. It results from primitive mesenchymal proliferation, and affects boys slightly more often than girls. Histologically, it is composed of disordered, primitive, fluid-filled mesenchyme, hepatic parenchyma, and bile ducts in addition to stromal cysts of variable size without a capsule. Alpha-fetoprotein levels are usually normal.
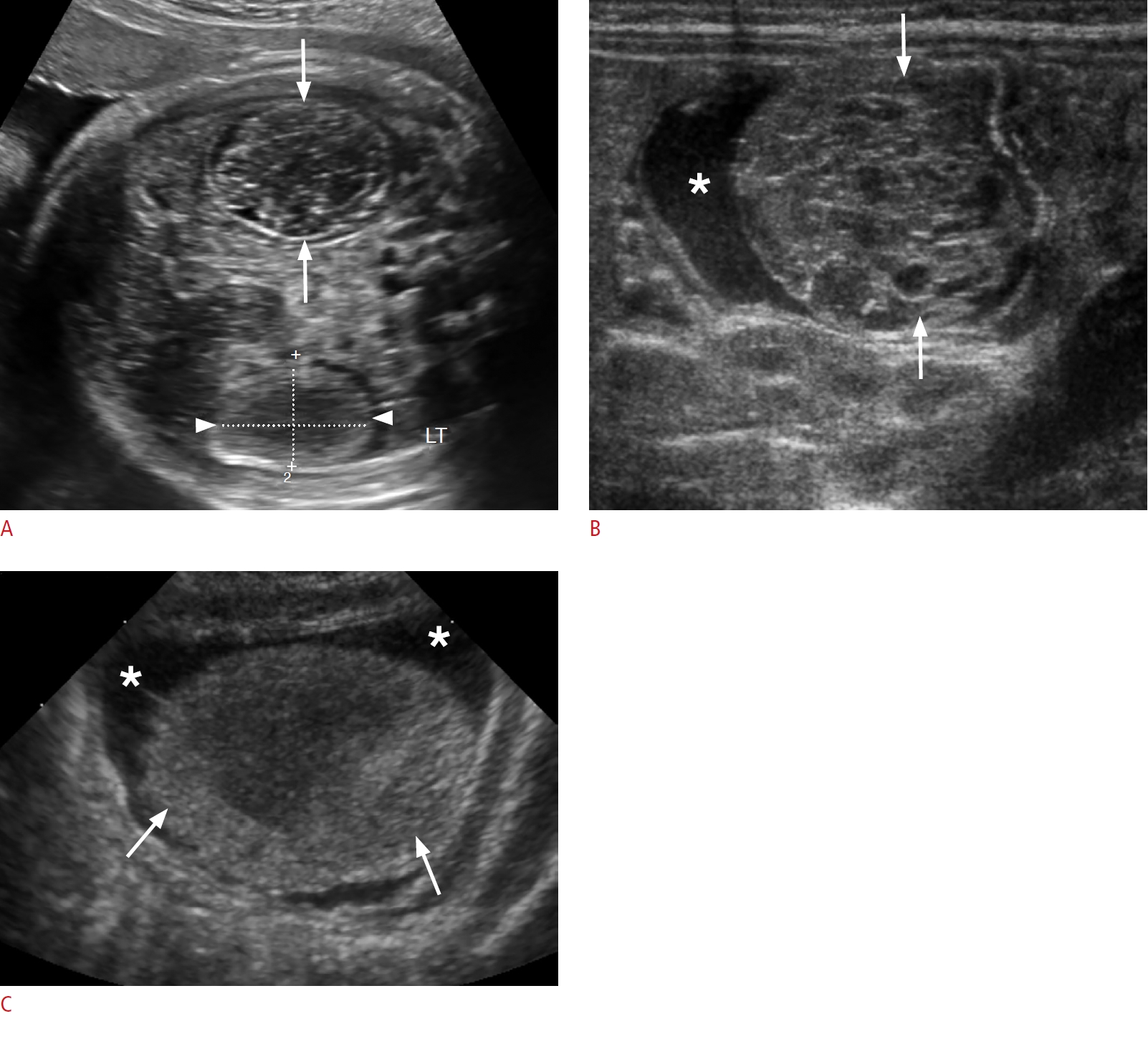
Mesenchymal hamartomas are typically multicystic, heterogeneous masses with septa of variable thickness, most commonly arising in the right lobe of the liver (Fig. 9A-C). They are typically unifocal and show little blood flow on a Doppler exam. When the cysts are tiny, the lesion is hyperechoic and simulates a solid lesion. Calcification and hemorrhage are uncommon. Low-level echoes may be seen within the cyst fluid related to gelatinous contents or hemorrhage. On MRI, mesenchymal hamartomas are complex cystic masses with variable signal intensity, related to the stromal content, amount of protein and presence or absence of hemorrhage within the cyst. The cystic regions are usually hypointense on T1 and hyperintense on T2-weighted images with enhancement of the septa and solid components (Fig. 9D, E). Mesenchymal hamartomas have an excellent prognosis. The treatment is surgical resection due to rare reports of malignant transformation to undifferentiated embryonal sarcoma [55-59].
Renal and Retroperitoneal Cystic Masses
Multicystic Dysplastic Kidney
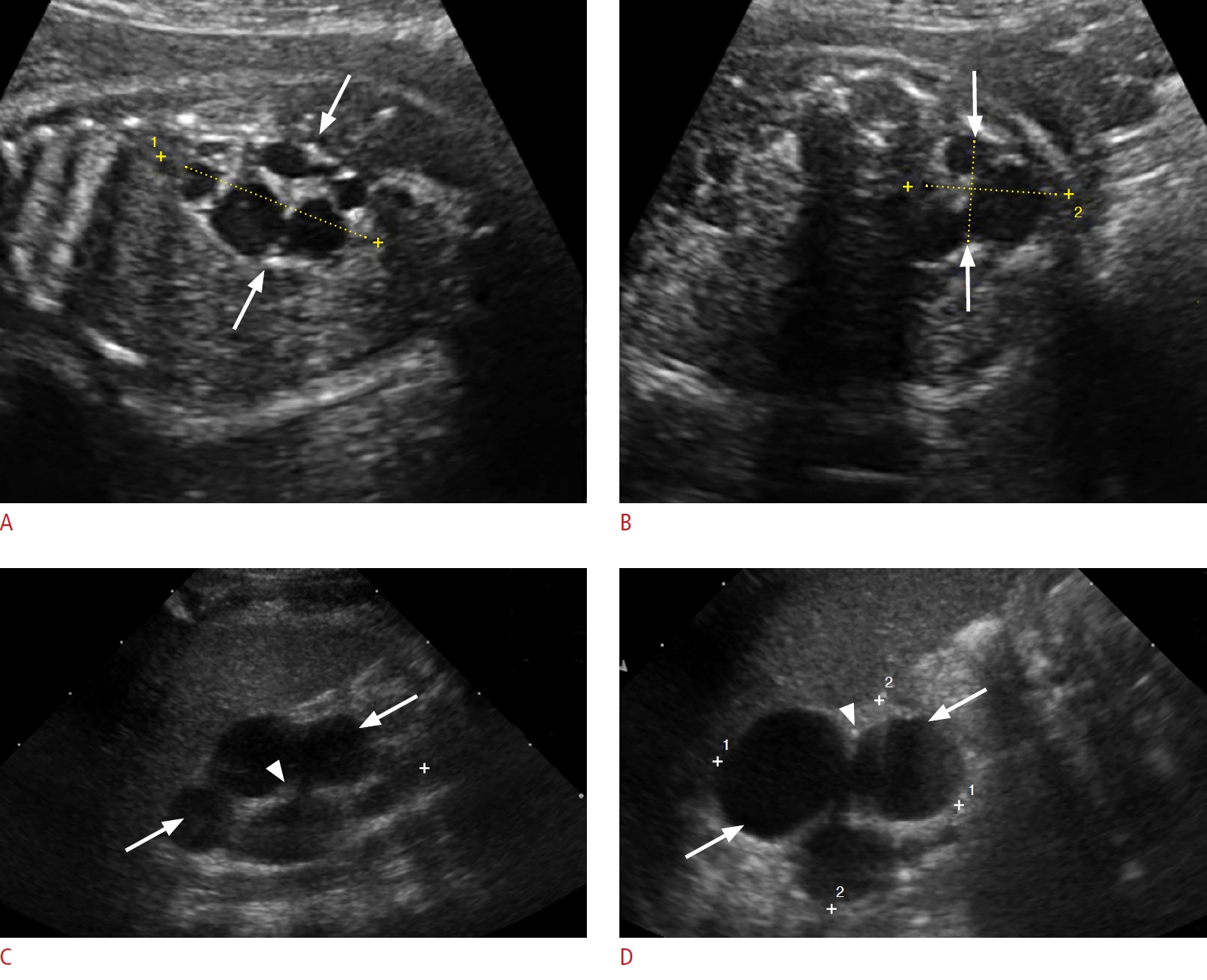
Multicystic dysplastic kidney (MCDK) is the most common cystic disorder of infants and children. It results from severe obstruction or atresia of the renal pelvis and/or ureter during early urogenital development, leading to disordered parenchymal development. Most cases are sporadic, but familial cases have been described [60,61]. The two forms of MCDK include the pelvoinfundibular type (common) and the hydronephrotic type (uncommon) [60]. MCDK can occur in half of a duplex kidney, horseshoe kidney, or crossed fused renal ectopia [62]. Vesicoureteral reflux and contralateral ureteropelvic junction (UPJ) obstruction can be associated with this anomaly. Müllerian anomalies may coexist in girls [62].
On imaging, the classic MCDK is characterized by multiple cysts of variable sizes that do not communicate, and poorly defined intervening echogenic renal parenchyma without normal corticomedullary architecture (Fig. 10). The cysts may communicate in the hydronephrotic form mimicking pelvicaliectasis, but with lack of normal renal parenchyma. The affected kidney may be small, normal or enlarged and compensatory hypertrophy of the contralateral kidney is often present. MCDK can be confirmed by renal scintigraphy or magnetic resonance urography (MRU), which document a characteristic absence of renal function [63].
The vast majority of MCDK involute with time and remain asymptomatic [64-66]. The treatment is follow-up; resection is rarely indicated and usually reserved for cases in which there is increasing kidney size rather than involution [60]. Unilateral MCDK with normal contralateral kidney has an excellent prognosis. Renal insufficiency, pyelonephritis, and scarring can be a problem in cases of unilateral MCDK with an abnormal contralateral kidney. Bilateral involvement is incompatible with life [61].
Urinoma
Perinatal urinoma is quite rare and presents as an encapsulated collection of urine within the perirenal fascia, caused by calyceal microperforation with leakage of urine resulting from severe renal obstruction. The most common causes of high-grade obstruction are posterior urethral valves or UPJ obstruction [67]. Upper urinary tract obstruction may be associated with a worse prognosis for the kidney than lower urinary tract obstruction [67-69]. Most urinomas are unilateral. The majority occur in boys [70].
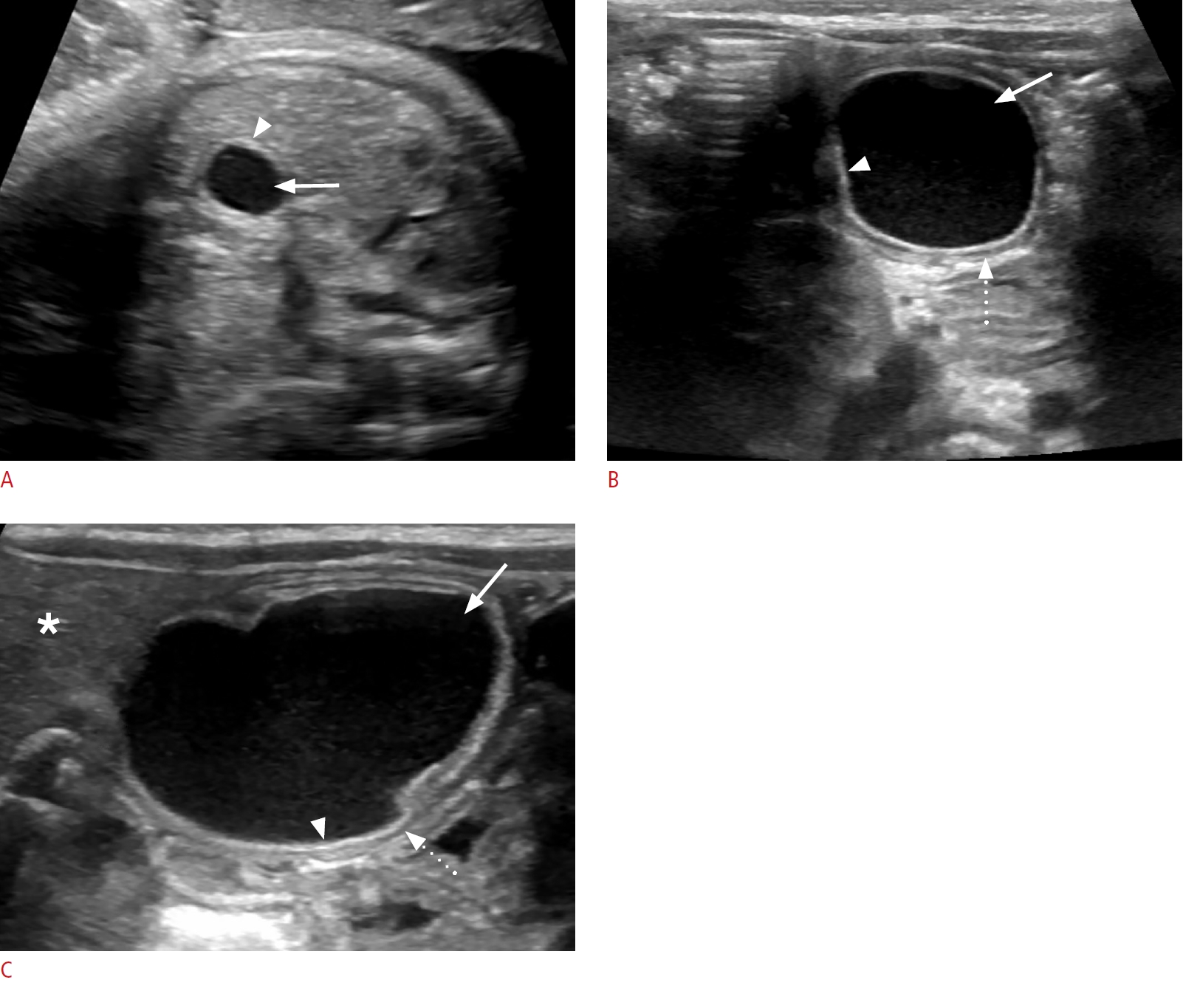
On sonography, a urinoma appears as crescentic, round, or elliptical anechoic fluid collection around or abutting an obstructed kidney. It is more often unilocular than septated, seen adjacent to the spine. The ipsilateral kidney may be distorted and displaced. Associated findings of ipsilateral severe renal obstruction are typically present, characterized by a variable amount of pelvicalyceal and urinary tract dilation and echogenic renal parenchyma (Fig. 11A, C). UPJ obstruction or dilated posterior urethra may be present (Fig. 11B) [71]. Nearly all kidneys with urinoma are subsequently shown to have decreased or no function. Spontaneous resolution of urinomas may occur, often reflecting oliguria of the affected kidney, and does not change the prognosis [67]. Management involves treatment of the underlying cause of the urinary tract obstruction [70].
Duplex Kidney with Severe Hydronephrosis Dysplastic Parenchyma of the Upper Moiety
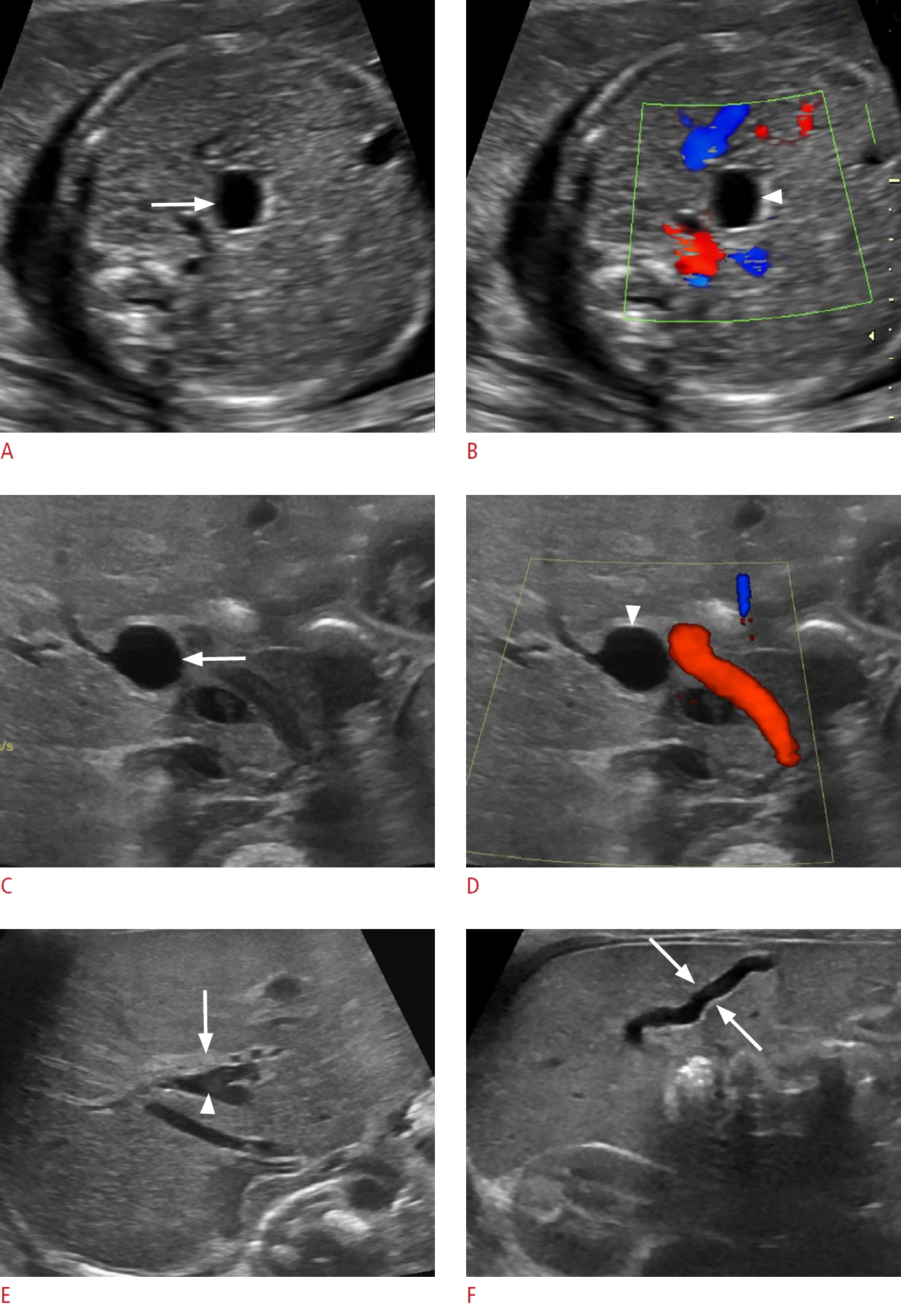
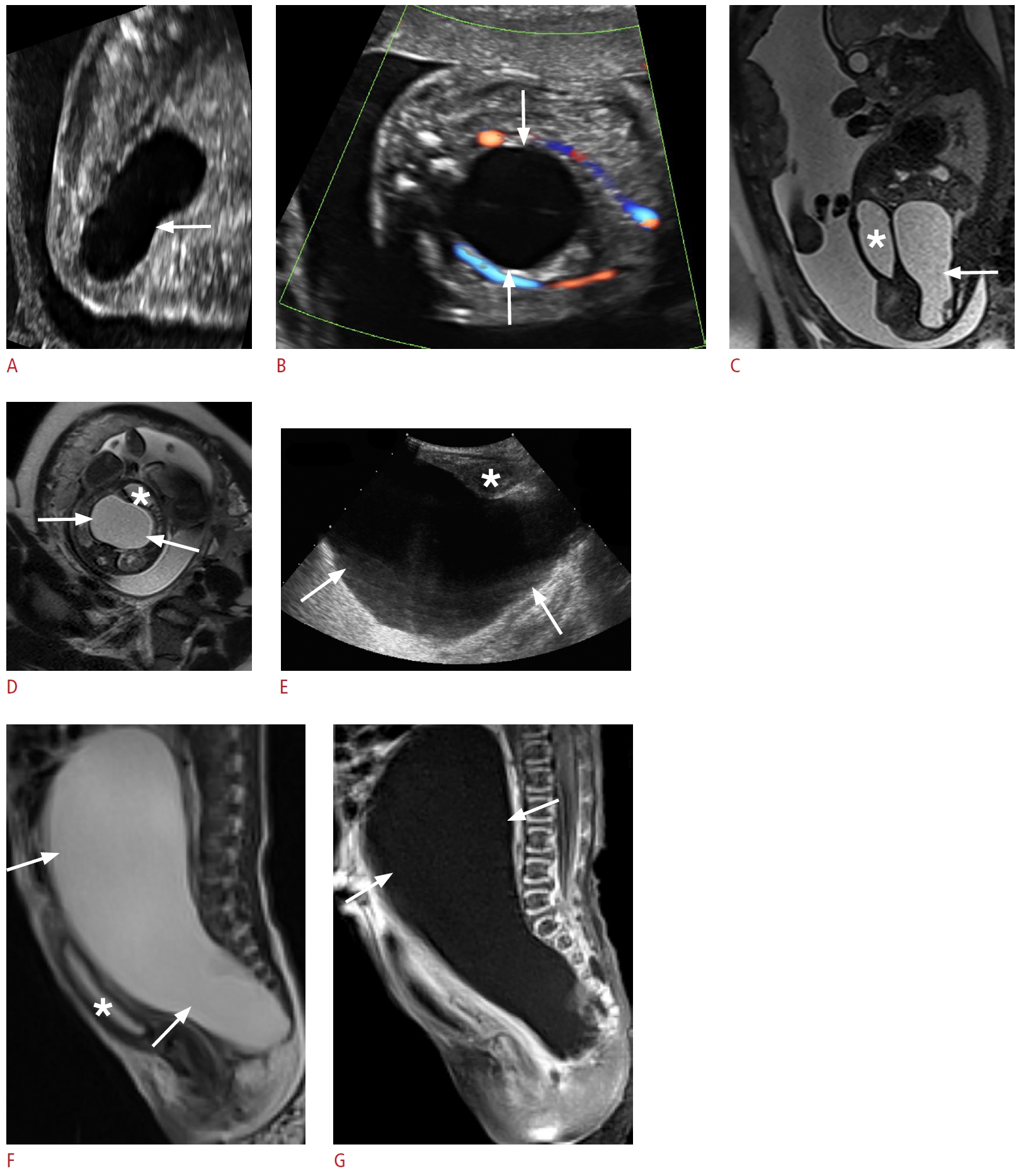
A duplex kidney consists of upper and lower renal moieties drained by separate collecting systems and/or ureters. It occurs when two ureteric buds are present embryologically, or the ureteric bud bifurcates before meeting the metanephric blastema [72,73]. Complete duplications with a separate ureter draining each collecting system are less common than incomplete duplications with conjoined draining ureters. The prevalence has been reported to be 0.7%-4%, with females affected more than males [73]. The left side is involved more often than the right and bilateral duplication occurs in about 20% to 40% of affected individuals. When complete, the upper moiety ureter inserts into the bladder wall inferior and medial to the lower moiety ureter and is more likely to be obstructed. In contrast, the lower moiety ureter is more likely to reflux [73,74]. Complete duplication can present with severe hydronephrosis or bladder outlet obstruction due to a large ectopic ureterocele (Fig. 12) [73,74]. Severe hydronephrosis of the upper moiety in a duplex kidney with dysplastic renal parenchyma can mimic a cystic suprarenal mass on sonography. The presence of a thin-walled intraluminal cystic lesion within the bladder representing an ectopic ureterocele, confirms that the cystic retroperitoneal mass is related to an obstructed upper renal moiety rather than an adrenal mass (Fig. 12C) [73,75]. Further evaluation of complicated duplex kidneys using voiding cystourethrography (VCUG), and at times MRU may be required [74]. Treatments for upper moiety obstruction include endoscopic puncture or excision of an obstructing ureterocele, ureteroureterostomy, and upper moiety heminephrectomy in the setting of poor or absent kidney function [76].
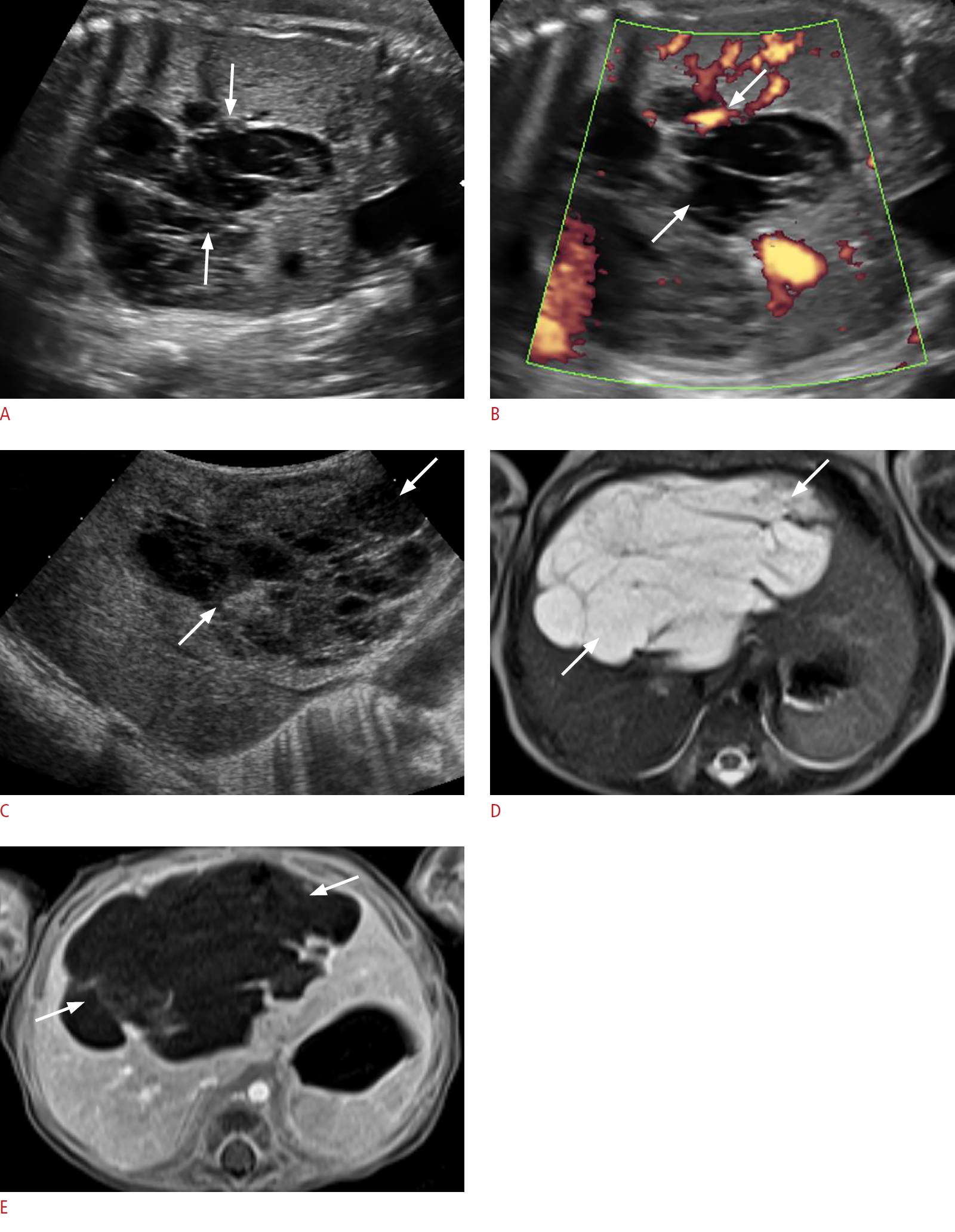
Cystic Neuroblastoma
Congenital neuroblastoma is the most common prenatally detected neonatal adrenal neoplasm, and is cystic in 25% of cases. It is frequently right-sided and may present as localized disease in the newborn or with metastases [71,77]. On sonography, cystic neuroblastoma appears as a complex cystic mass with thick septations and internal blood flow on color Doppler imaging (Fig. 13). Absence of blood flow, however, does not exclude the diagnosis of neuroblastoma. The sonographic findings of cystic neuroblastoma may overlap with those of adrenal hemorrhage; however, the latter uncommonly presents prenatally. In addition, on follow-up ultrasonography, adrenal hemorrhage typically decreases in size and changes echogenicity within the first week and eventually resolves after several weeks, often leaving a residual focus of calcification. In contrast, sonographic appearance changes less rapidly in cystic neuroblastoma and calcification is less common [71]. MRI may be useful for confirming the anatomical origin and for staging and evaluating metastases [71]. Most congenital neuroblastomas have a good prognosis; they commonly have a favorable stage (L1, L2, and MS) and biologic markers. They may resolve spontaneously or remain stable without complications. Surgery may be curative in some cases [59,78].
Pancreatic Cysts
Congenital pancreatic cysts are rarely encountered; they possess a true epithelium and are thought to arise from a developmental anomaly of the pancreatic ductal system [79]. They may be isolated or associated with systemic diseases such as Beckwith-Wiedemann syndrome, polycystic renal disease, von Hippel-Lindau syndrome, asphyxiating thoracic dystrophy, and heterotaxy syndrome [79-83]. They are small and anechoic on sonography, unilocular or multilocular, and mostly located in the pancreatic body or tail. They are surrounded by normal parenchyma and rarely communicate with the ductal system. Complications include cyst infection, inflammation, or rupture; cholangitis; pancreatitis; and peritonitis [18,80]. Surgical removal is only necessary for symptomatic cysts or those concerning for neoplasia [79-81].
Splenic Cysts
Congenital splenic cysts are rare. They are true cysts with an epithelial lining [84]. The pathogenesis is unclear, although these cysts have been proposed to arise from production of fluid from pluripotent cells or from the normal lymph spaces in the spleen [85]. Congenital splenic cysts are typically isolated and asymptomatic, and they rarely develop complications [84,86]. They are small, anechoic on sonography, with a smooth imperceptible wall, no mural calcification, and no color Doppler flow. The surrounding parenchyma is usually normal [85]. Most congenital splenic cysts remain stable, decrease in size, or completely regress [85-87]. Although rare, sporadic cases of rupture, hemorrhage, and infection have been reported in the literature [88,89]. Symptomatic and large congenital splenic cysts may require percutaneous drainage and/or sclerotherapy. Total or partial splenectomy and partial cystectomy with marsupialization or cyst fenestration have been described [84].
Cystic Masses Arising from the Pelvis
Hydrocolpos and Hydrometrocolpos
Congenital hydrocolpos is uncommon, characterized by fluid distention of the vagina, secondary to vaginal tract obstruction. Hydrometrocolpos refers to fluid accumulation in the obstructed vagina and uterus. It is believed to result from increased mucus secretion by the vagina and cervix in response to maternal hormones; the mucus gradually accumulates, expands, and builds up into a pelvic mass due to vaginal obstruction. Persistent urogenital sinus and cloacal dysgenesis are commonly associated with congenital hydrocolpos; mixing of urine, vaginal secretions, and meconium will be seen in this context. Vaginal tract obstruction can also occur from vaginal atresia, transverse vaginal septum, and imperforate hymen. Most cases of congenital hydrocolpos are sporadic, but this condition can be associated with several syndromes [90,91].
On sonography, hydrocolpos appears as a unilocular, fluid-filled pelvic mass posterior to the bladder with characteristic funneling to the perineum. The cervix may be opened with contiguous fluid distending the uterine cavity, usually of lesser magnitude than the vagina because of the thick muscular uterine wall. Fluid-fluid level or mobile echoes may be seen within the cystic mass (Fig. 14). Further imaging with a genitogram, VCUG, and MRI is usually required in patients with coexisting genitourinary anomalies [90-93].
The most common complication of congenital hydrocolpos is hydronephrosis and renal failure. Intestinal obstruction, cardiorespiratory distress, and sepsis can also occur [90,91,94]. The initial management is often urgent percutaneous drainage, followed by surgical management, aimed at correcting the obstruction [90-92].
Cystic Sacrococcygeal Teratoma
Sacrococcygeal teratoma is the most common congenital neoplasm of neonates, with a female preponderance of 4:1 [59,95]. The tumors may be purely cystic in 15% of cases. Teratomas are composed of all three germ cell layers (ectoderm, mesoderm, and endoderm). They commonly occur along the midline of the body, anywhere from the coccyx to the pineal gland. The majority of teratomas arise from the sacrococcygeal region, and less frequently in the retroperitoneum [95-97]. Sacrococcygeal teratomas are classified according to the American Academy of Pediatrics Surgery Section Survey into four types (I-IV) based on the amount of mass present externally versus internally, which has important prognostic implications. Cystic teratomas are anechoic on sonography without internal blood flow. In type I, the cystic mass is predominantly external; in type II, there is extension to the presacral space; in type III, the mass extends into the abdominal cavity; and in type IV, the mass is entirely internal [59,98-100]. Further MRI is useful to assess the size, characteristics, and extent of the mass to optimize management (Fig. 15) [95,97]. Associated conditions may include hydronephrosis, renal dysplasia, urethral atresia, urinary ascites, hydrocolpos, and undescended testes [59,96,99,100]. Predominantly cystic tumors have a better prognosis, presumably because of the lower prevalence of vascular steal and hemorrhage. The treatment for sacrococcygeal teratoma is complete resection [95,97,100].
Conclusion
Prenatally detected abdominal and pelvic cystic lesions can arise from many structures in the abdomen and have a broad differential diagnosis. Ultrasonography should be the first imaging investigation following birth and is often the only imaging study required to arrive at a diagnosis, supplemented by MRI when necessary. An organized anatomical approach and attention to characteristic sonographic findings aid diagnosis and guide appropriate management.
 Download Citation
Download Citation PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC



